Spletna revija za znanstvenike, strokovnjake
in nevroznanstvene navdušence
Naslovnica Članki Intervjuji Mnenja Zdravje Korenine eSinapsa Številke ![]()
Bolezni spektra anti-MOG pri odraslih
letnik 2020, številka 18
uvodnik
Maša Čater
članki
Bolezni spektra anti-MOG pri odraslih
Nik Krajnc
Samomor pod lupo nevroznanosti
Alina Holnthaner
intervjuji
Miha Pribošič
aktualno
Preplet možganov in imunskega sistema v času stresa
Dolores Trol
kolofon
letnik 2020, številka 18
Bolezni spektra s protitelesi proti mielinskemu oligodendrocitemu glikoproteinu (MOG) predstavljajo redko demielinizacijsko obolenje osrednjega živčevja. Pri odraslih najpogosteje poteka kot optični nevritis in/ali transverzni mielitis. Optični nevritis se kaže s poslabšanjem ostrine vida na prizadeto oko ter bolečino ob premikanju zrkel, bolniki pa tožijo tudi za slabšim zaznavanjem barv, predvsem rdeče. Transverzni mielitis predstavlja vnetje hrbtenjače, ki se kaže z raznoliko klinično sliko, vključno z mišično šibkostjo v spodnjih okončinah, spremenjenim zaznavanjem dotika, vibracij, bolečine in temperature ter motnjami odvajanja vode in blata. Ob pojavu bolezni bolniki prejmejo pulzno zdravljenje s kortikosteroidi, potrebujejo pa tudi vzdrževalno zdravljenje (azatioprin, rituksimab). Ključno je razlikovanje z multiplo sklerozo, saj lahko nekatera izmed imunomodulatornih zdravil neugodno vplivajo na potek bolezni spektra anti-MOG.
Uvod
Med vnetne demielinizacijske bolezni osrednjega živčevja poleg multiple skleroze (MS) uvrščamo tudi druga demielinizacijska obolenja, vključno z boleznimi spektra nevromielitis optika (angl. neuromyelitis optica spectrum disorders, NMOSD) in akutnim diseminiranim encefalomielitisom (ADEM). Zgodnje razlikovanje med njimi je ključno za oblikovanje ustreznih terapevtskih pristopov, vendar je zaradi prekrivajočih se kliničnih in parakliničnih značilnosti pogosto oteženo.
Leta 2004 so bila pri bolnikih z NMOSD prvič opisana protitelesa, usmerjena proti vodnemu kanalčku akvaporinu 4 (angl. aquaporin 4, AQP4), ki je bogato zastopan na astrocitnih podaljških 1, 2. V živalskih modelih MS (eksperimentalni avtoimunski encefalomielitis, EAE) predstavlja eno izmed tarčnih struktur tudi mielinski oligodendrocitni glikoprotein (MOG) 3, 4. Približno tri četrtine bolnikov z NMOSD ima anti-AQP4, med preostalimi pa anti-MOG najdemo le v 20–25 %. V značilno klinično sliko bolezni spektra anti-MOG uvrščamo rekurentni optični nevritis (ON), redkeje transverzni mielitis, ki je pogostejši pri anti-AQP4 pozitivnih NMOSD 5. V zadnjih letih se je močno razširilo znanje o boleznih spektra anti-MOG, vključno s kliničnimi, radiološkimi in laboratorijskimi najdbami, ki jih podajamo v nadaljevanju.
Patofiziologija
MOG se nahaja na mielinski ovojnici in površini oligodendrocitnih izrastkov v osrednjem živčevju (slika 1) in je neposredno imunogen 6, 7. Anti-MOG so imunoglobulini (Ig) razreda G, ki povzročijo od komplementa odvisno citotoksičnost 8. Nastanejo na periferiji in dosežejo osrednji živčni sistem ob porušeni krvno-možganski pregradi, npr. zaradi okužbe 9. Odsotnost oligoklonalnih trakov, ki predstavljajo nespecifičen označevalec vnetja v osrednjem živčevju, in anti-MOG v likvorju podpira hipotezo o perifernem izvoru sindroma 10.
Patološke raziskave v področju demielinizacijskih lezij razkrivajo depozite IgG in aktivacijo komplementa, kar srečamo tudi pri bolnikih z MS 11, 12. Lezije so povezane z izgubo mielina z relativno ohranjenimi aksoni in astrociti, v okolici pa se nahajajo makrofagi z ostanki mielina. Vnetni infiltrati se nahajajo ob žilah in vsebujejo predvsem limfocite T 11. Shematski prikaz patofiziologije bolezni spektra anti-MOG prikazuje slika 1 13.
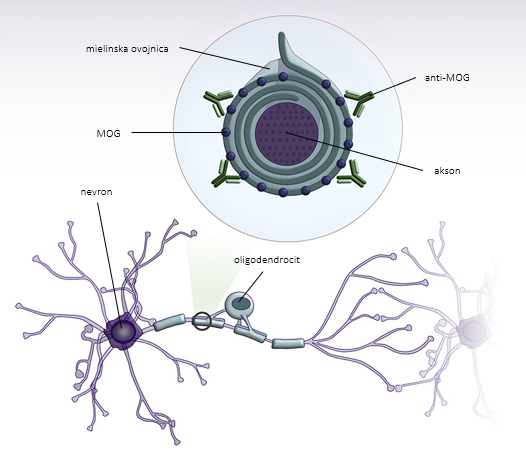
Slika 1. Shematski prikaz patofiziologije bolezni spektra anti-MOG. Protitelesa anti-MOG so usmerjena proti mielinskemu oligodendrocitnemu glikoproteinu (MOG), ki se nahaja na površini mielinske ovojnice. Prirejeno po Pérez et al., 2019 13.
Diagnostična merila
Leta 2018 so bila objavljena diagnostična merila za bolezni spektra anti-MOG pri odraslih 14. Za diagnozo je potrebno izpolniti vsa merila 14:
• monofazni ali rekurentni ON, mielitis, encefalitis možganskega debla ali katera koli kombinacija navedenih simptomov,
• izvid MRI ali elektrofizioloških preiskav (npr. vidni izvabljeni odzivi pri bolnikih z izoliranim ON), skladen z demielinizacijo osrednjega živčevja, in
• pozitivni anti-MOG v serumu, dokazani na celični liniji.
Težavnost diagnostike bolezni spektra anti-MOG pa nastopi v naslednjih primerih. Kroničen napredujoč potek bolezni (napredujoče oblike MS, sarkoidoza, tumorji), klinične in paraklinične najdbe, ki govorijo v prid druge diagnoze (tuberkuloza, borelioza, sifilis, Behcetova bolezen, subakutna kombinirana degeneracija, limfom, paraneoplastična obolenja), in nizek titer anti-MOG predstavljajo najdbe, neznačilne za bolezni spektra anti-MOG (angl. red flags), ki kažejo na obstoj drugih diferencialno diagnostičnih možnostih 10.
Klinična slika
Prvič so bila protitelesa anti-MOG opisana leta 2007 pri ADEM, ki predstavlja najpogostejšo obliko bolezni spektra anti-MOG pri otrocih, medtem ko ima večina (80 %) odraslih bolnikov polifazen potek 5, 15. Najpogosteje poteka kot izoliran ON ali kombiniran ON in mielitis, izoliran mielitis srečamo redkeje 16. Dva- do trikrat pogosteje prizadene ženske, večinoma v starosti od 31 do 37 let 17, 18, 19. K predispoziciji žensk k avtoimunskim boleznim najbolj prispevajo razlike v spolnih hormonih 20, 21, 22.
Bolezen se po osmih letih ponovno pojavi pri 93 % bolnikov, pri čemer se največkrat kaže s sliko ON (88 %), mielitisa (56 %) ali prizadetosti možganskega debla (24 %), redkeje kot encefalitis (14 %) ali cerebelitis (4 %) 23. Opisani so tudi sindromi prekrivanja z avtoimunskim encefalitisom s protitelesi proti receptorju za N-metil-D-aspartat (anti-NMDAR) in ON, povezanim s protitelesi proti domeni α1 glicinskega receptorja (anti-GlyR) 24, 25.
ON je v primerjavi z drugimi demielinizacijskimi obolenji pogosteje obojestranski 26. Obojestransko prizadetost je dokazala tudi raziskava Havle in sodelavcev, v kateri so pri bolnikih z enostranskim ON tudi na asimptomatskem očesu dokazali atrofijo retinalnih vlaken 27.
Bolezni spektra anti-MOG so za razliko z anti-AQP4 NMOSD pogosteje pridruženi encefalopatija, retrogradna amnezija in epileptični napadi 28, 29. Razlikujeta se tudi po okrevanju po zagonu bolezni, ki je boljše pri bolnikih z anti-MOG z nižjo oceno prizadetosti po Razširjeni lestvici stopnje prizadetosti (angl. Expanded Disability Status Score, EDSS) 30, 31, 32.
Diagnostika
Laboratorijske preiskave
Laboratorijska diagnostika degenerativnih bolezni osrednjega živčevja najpogosteje temelji na pregledu hrbtenjačnega likvorja. Značilna protitelesa anti-MOG najdemo le v serumu, dokazov o sintezi znotraj osrednjega živčevja le-teh ni 33. Kljub temu v 55–70 % primerov v likvorskem izvidu ugotavljamo povečano število limfocitov, število celic pa je praviloma višje v primerjavi z MS 5, 30, 31, 33. Oligoklonalni trakovi so pozitivni v 10 % 33, 34.
Nizke vrednosti anti-MOG najdemo tudi pri zdravih preiskovancih in bolnikih z drugimi nevrološkimi obolenji, zato je bil sprva za spodnjo mejo določen titer ≥ 1:160. Kasneje so za mejno vrednost predlagali titer ≥ 1:1.280, ki omogoča večjo specifičnost preiskave z nekoliko nižjo občutljivostjo 35, 36.
Slikovna diagnostika
Na MRI glave pri dveh tretjinah bolnikov ne ugotavljamo posebnosti oz. najdemo le nespecifične subkortikalne lezije. Drobne lezije so lahko prisotne tudi v globoki beli možganovini in so večinoma asimptomatske 37. V redkih primerih imajo lezije predilekcijo za infratentorialne predele, predvsem mezencefalon, pons, olivi in cerebelarne pedunkle 38, 39. Lezije imajo puhast (angl. fluffy) izgled za razliko od za MS značilnih ovoidnih lezij 40. Razlikovanje med bolniki z anti-AQP4 in anti-MOG je na MRI glave precej težavno. Pri slednjih lezije pogosteje najdemo v talamusu in ponsu, vendar omenjene najdbe niso specifične 28.
ON zajema pri bolnikih z anti-MOG sprednje segmente vidnega živca, vključno z intraorbitalnim delom, vidno pa je tudi perinevralno ojačanje signala 38, 39. Optična kjazma značilno ni prizadeta 41. Obojestranski ON se pojavi v četrtini primerov. Nasprotno ON pri bolnikih z anti-AQP4 praviloma prizadene posteriorne segmente, vključno z optično kjazmo 39. Slika 2 prikazuje značilen obojestranski ON, povezan z boleznijo spektra anti-MOG.
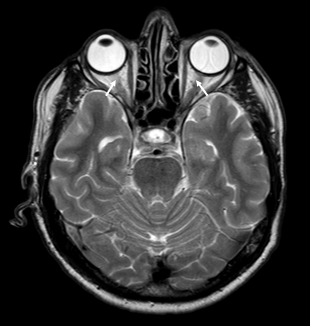
Slika 2. Slika 2. Optični nevritis (ON), povezan z anti-MOG, značilno prizadene intraorbitalni del optičnih živcev (označeno s puščico), medtem ko optična kjazma praviloma ni prizadeta.
V sklopu mielitisa ločimo dva različna vzorca prizadetosti hrbtenjače, in sicer dolge lezije, ki se raztezajo prek treh ali več vretenc (angl. longitudinally ectensive transvense myelitis, LETM) in zajamejo vsaj polovico preseka hrbtenjače, ter kratke lezije, krajše od dveh vretenc 33, 39. Mielitis se najpogosteje pojavi v torakolumbalnem predelu za razliko od cervikotorakalne prizadetosti pri bolnikih z anti-AQP4 37. Tudi lezije v konusu so visoko specifične za bolezni spektra anti-MOG 30.
Tabela 1 predstavlja ključne klinične in paraklinične razlike med MS, NMOSD in boleznimi spektra anti-MOG.
Tabela 1. Ključne klinične in paraklinične razlike med MS, NMOSD in boleznimi spektra anti-MOG 40, 51. OGT – oligoklonalni trakovi.
| MS | NMOSD | Anti-MOG | ||
|---|---|---|---|---|
| razmerje ženske:moški | 3:1 | 9:1 | 3:1 | |
| starost ob nastopu bolezni | 20–30 let | 35–45 let | 31–37 let | |
| serumska protitelesa | brez | Anti-AQP4 v 60–90 %, anti-MOG pri 20–25 % anti-AQP4 negativnih bolnikov | anti-MOG | |
| potek ON | monofazni, redko bilateralni | monofazni ali rekurentni, pogosto bilateralni, nepopolno okrevanje | rekurentni, pogosto bilateralni | |
| prizadetost hrbtenjače (mielitis) | pogosto | pogosto | redkeje | |
| druge manifestacije | area postrema sindrom, sindrom možganskega debla, simptomatska narkolepsija, sindrom diencefalona | |||
| likvorski izvid | OGT | brez OGT, redko anti-AQP4 | brez OGT, redko anti-MOG | |
| izvid OCT | minimalna izguba ganglijskih celic, homogena distribucija okoli makule | pomembna izguba ganglijskih celic, lokalizirano fovealno in parafovealno | ||
| slikovna diagnostika (MRI) | vidni živec | sprednji del vidnega živca, krajši segment | zadnji del vidnega živca do sprednjega dela kjazme, daljši segment | sprednji del vidnega živca, daljši segment |
| možgani | pogosto; ovoidne lezije z dobro definiranim robom (periventrikularno, kortikalno ali jukstakortikalno, infratentorialno) | občasno; puhaste lezije (area postrema, talamus, hipotalamus, zdajšnji del hipofize, pinealna žleza) | redko; puhaste lezije (pons, periventrikularno) | |
| hrbtenjača | pogosto; vratni del hrbtenjače, številne kratke lezije posterolateralno | pogosto; centralne lezije cervikotorakalno, LETM | redkeje; centralne ali lateralne lezije torakolumbalno, 67 % LETM, preostalo kratke lezije |
Zdravljenje
Smernic za zdravljenje bolnikov z boleznimi spektra anti-MOG še nimamo, zato se opiramo na smernice za obravnavo bolnikov z NMOSD 42, 43. Ključno je razlikovanje z MS, saj lahko nekatera izmed imunomodulatornih zdravil, ki jo uporabljamo za zdravljenje MS (interferon β, glatiramer acetat, dimetilfumarat, fingolimod, natalizumab, alemtuzumab), negativno vplivajo na potek bolezni 5,44.
Ob zagonu bolniki prejmejo tri- ali petdnevno pulzno zdravljenje v odmerku 1 g metilprednizolona na dan 45. Glede na jakost zagona se lahko po pulznem zdravljenju odločimo tudi za peroralno zdravljenje s kortikosteroidi v padajočem odmerku 46. Bolnike s hujšimi zagoni ali suboptimalnim odgovorom na zdravljenje s kortikosteroidi zdravimo s plazmaferezo, redkeje z intravenskimi imunoglobulini 10. Plazmafereza je postopek, analogen dializi, s katerim bolniku odstranimo del krvne plazme, jo prečistimo (odstranimo imunoglobuline in preostale komponente krvi, razen krvnih celic) ter vrnemo v krvni obtok.
Bolniki potrebujejo tudi dolgotrajno vzdrževalno zdravljenje z imunosupresivno terapijo. Večina bolnikov prejema azatioprin ali rituksimab, kot zdravilo drugega izbora pa pridejo v poštev tudi mikofenolat mofetil, mitoksantron in metotreksat 9, 43. Azatioprin ostaja najpogosteje uporabljeno zdravilo za preprečevanje zagonov bolezni, ob katerem je ključno redno spremljanje hemograma in testov jetrne funkcije, med pogoste neželene učinke pa uvrščamo tudi slabost in bruhanje 47, 48.
Za preprečevanje zagonov NMOSD je od junija 2019 na voljo tudi ekulizumab (anti-C5), vendar podatkov o uspešnosti preprečevanja zagonov pri bolnikih z anti-MOG zaenkrat še ni 49. V fazi kliničnih preizkušanj je trenutno tudi inebilizumab (anti-CD19) 50.
Zaključek
Bolezni spektra anti-MOG predstavljajo redko demielinizacijsko obolenje osrednjega živčevja, ki kljub nekaterim vzporednicam z NMOSD predstavlja svojevrstno klinično entiteto. Diagnoza temelji na značilni klinični sliki in izvidih parakliničnih preiskav, pri čemer je ključna določitev titra protiteles anti-AQP4 in anti-MOG v serumu. Zaradi relativno nizke incidence bolezni spektra anti-MOG smernic za obravnavo tovrstnih bolnikov še nimamo, zato se opiramo na smernice za zdravljenje bolnikov z NMOSD. Nadaljnje raziskave bodo pokazale, ali je patofiziološko razlikovanje z NMOSD z vidika terapevtske obravnave bolnikov z boleznimi spektra anti-MOG sploh smiselno.
-
___
-
Lennon, V.A., et al., A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet, 2004. 364(9451): p. 2106-12. ↩
-
Lennon, V.A., et al., IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med, 2005. 202(4): p. 473-7. ↩
-
Lebar, R., et al., Studies on autoimmune encephalomyelitis in the guinea pig. II. An in vitro investigation on the nature, properties, and specificity of the serum-demyelinating factor. J Immunol, 1976. 116(5): p. 1439-46. ↩
-
Kroepfl, J.F., et al., Investigation of myelin/oligodendrocyte glycoprotein membrane topology. J Neurochem, 1996. 67(5): p. 2219-22. ↩
-
Jarius, S., et al., MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflammation, 2016. 13(1): p. 280. ↩
-
Etemadifar, M., et al., Comparing myelin oligodendrocyte glycoprotein antibody (MOG-Ab) and non MOG-Ab associated optic neuritis: Clinical course and treatment outcome. Mult Scler Relat Disord, 2019. 27: p. 127-130. ↩
-
Kezuka, T. and H. Ishikawa, Diagnosis and treatment of anti-myelin oligodendrocyte glycoprotein antibody positive optic neuritis. Jpn J Ophthalmol, 2018. 62(2): p. 101-108. ↩
-
Mader, S., et al., Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflammation, 2011. 8: p. 184. ↩
-
Ramanathan, S., et al., Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry, 2018. 89(2): p. 127-137. ↩
-
Lana-Peixoto, M.A. and N. Talim, Neuromyelitis Optica Spectrum Disorder and Anti-MOG Syndromes. Biomedicines, 2019. 7(2). ↩
-
Spadaro, M., et al., Histopathology and clinical course of MOG-antibody-associated encephalomyelitis. Ann Clin Transl Neurol, 2015. 2(3): p. 295-301. ↩
-
Di Pauli, F., et al., Fulminant demyelinating encephalomyelitis: Insights from antibody studies and neuropathology. Neurol Neuroimmunol Neuroinflamm, 2015. 2(6): p. e175. ↩
-
Perez, C.A., et al., Overlapping autoimmune syndrome: A case of concomitant anti-NMDAR encephalitis and myelin oligodendrocyte glycoprotein (MOG) antibody disease. J Neuroimmunol, 2020. 339: p. 577124. ↩
-
Jarius, S., et al., MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflammation, 2018. 15(1): p. 134. ↩
-
O’Connor, K.C., et al., Self-antigen tetramers discriminate between myelin autoantibodies to native or denatured protein. Nat Med, 2007. 13(2): p. 211-7. ↩
-
Soelberg, K., et al., A population-based prospective study of optic neuritis. Mult Scler, 2017. 23(14): p. 1893-1901. ↩
-
Sepulveda, M., et al., Clinical spectrum associated with MOG autoimmunity in adults: significance of sharing rodent MOG epitopes. J Neurol, 2016. 263(7): p. 1349-60. ↩
-
Weber, M.S., et al., Defining distinct features of anti-MOG antibody associated central nervous system demyelination. Ther Adv Neurol Disord, 2018. 11: p. 1756286418762083. ↩
-
Di Pauli, F. and T. Berger, Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disorders: Toward a New Spectrum of Inflammatory Demyelinating CNS Disorders? Front Immunol, 2018. 9: p. 2753. ↩
-
Voskuhl, R., Sex differences in autoimmune diseases. Biol Sex Differ, 2011. 2(1): p. 1. ↩
-
Ngo, S.T., F.J. Steyn, and P.A. McCombe, Gender differences in autoimmune disease. Front Neuroendocrinol, 2014. 35(3): p. 347-69. ↩
-
Desai, M.K. and R.D. Brinton, Autoimmune Disease in Women: Endocrine Transition and Risk Across the Lifespan. Front Endocrinol (Lausanne), 2019. 10: p. 265. ↩
-
Jarius, S., et al., Screening for MOG-IgG and 27 other anti-glial and anti-neuronal autoantibodies in ‘pattern II multiple sclerosis’ and brain biopsy findings in a MOG-IgG-positive case. Mult Scler, 2016. 22(12): p. 1541-1549. ↩
-
Rojc, B., B. Podnar, and F. Graus, A case of recurrent MOG antibody positive bilateral optic neuritis and anti-NMDAR encephalitis: Different biological evolution of the two associated antibodies. J Neuroimmunol, 2019. 328: p. 86-88. ↩
-
Martinez-Hernandez, E., et al., Antibodies to aquaporin 4, myelin-oligodendrocyte glycoprotein, and the glycine receptor alpha1 subunit in patients with isolated optic neuritis. JAMA Neurol, 2015. 72(2): p. 187-93. ↩
-
Ramanathan, S., et al., Antibodies to myelin oligodendrocyte glycoprotein in bilateral and recurrent optic neuritis. Neurol Neuroimmunol Neuroinflamm, 2014. 1(4): p. e40. ↩
-
Havla, J., et al., Myelin-oligodendrocyte-glycoprotein (MOG) autoantibodies as potential markers of severe optic neuritis and subclinical retinal axonal degeneration. J Neurol, 2017. 264(1): p. 139-151. ↩
-
Cobo-Calvo, A., et al., Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults: The MOGADOR study. Neurology, 2018. 90(21): p. e1858-e1869. ↩
-
Hamid, S.H.M., et al., Seizures and Encephalitis in Myelin Oligodendrocyte Glycoprotein IgG Disease vs Aquaporin 4 IgG Disease. JAMA Neurol, 2018. 75(1): p. 65-71. ↩
-
Kitley, J., et al., Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol, 2014. 71(3): p. 276-83. ↩
-
Cobo-Calvo, A., et al., Antibodies to myelin oligodendrocyte glycoprotein in aquaporin 4 antibody seronegative longitudinally extensive transverse myelitis: Clinical and prognostic implications. Mult Scler, 2016. 22(3): p. 312-9. ↩
-
Chen, J.J., et al., Myelin Oligodendrocyte Glycoprotein Antibody-Positive Optic Neuritis: Clinical Characteristics, Radiologic Clues, and Outcome. Am J Ophthalmol, 2018. 195: p. 8-15. ↩
-
Jarius, S., et al., MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 1: Frequency, syndrome specificity, influence of disease activity, long-term course, association with AQP4-IgG, and origin. J Neuroinflammation, 2016. 13(1): p. 279. ↩
-
Jurynczyk, M., et al., Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain, 2017. 140(12): p. 3128-3138. ↩
-
Peschl, P., et al., Myelin Oligodendrocyte Glycoprotein: Deciphering a Target in Inflammatory Demyelinating Diseases. Front Immunol, 2017. 8: p. 529. ↩
-
Hennes, E.M., et al., Prognostic relevance of MOG antibodies in children with an acquired demyelinating syndrome. Neurology, 2017. 89(9): p. 900-908. ↩
-
Sato, D.K., et al., Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology, 2014. 82(6): p. 474-81. ↩
-
Ramanathan, S., et al., Radiological differentiation of optic neuritis with myelin oligodendrocyte glycoprotein antibodies, aquaporin-4 antibodies, and multiple sclerosis. Mult Scler, 2016. 22(4): p. 470-82. ↩
-
Deneve, M., et al., MRI features of demyelinating disease associated with anti-MOG antibodies in adults. J Neuroradiol, 2019. 46(5): p. 312-318. ↩
-
Jurynczyk, M., et al., Brain lesion distribution criteria distinguish MS from AQP4-antibody NMOSD and MOG-antibody disease. J Neurol Neurosurg Psychiatry, 2017. 88(2): p. 132-136. ↩
-
Biotti, D., et al., Optic neuritis in patients with anti-MOG antibodies spectrum disorder: MRI and clinical features from a large multicentric cohort in France. J Neurol, 2017. 264(10): p. 2173-2175. ↩
-
Caron-Cantin, M., D.M. Cestari, and E. Fortin, Clinical and radiologic approach to ‘typical’ versus antibody-related optic neuritis. Curr Opin Ophthalmol, 2019. 30(6): p. 412-417. ↩
-
Sellner, J., et al., EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol, 2010. 17(8): p. 1019-32. ↩
-
Trebst, C., et al., Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the Neuromyelitis Optica Study Group (NEMOS). J Neurol, 2014. 261(1): p. 1-16. ↩
-
Hacohen, Y., et al., Disease Course and Treatment Responses in Children With Relapsing Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease. JAMA Neurol, 2018. 75(4): p. 478-487. ↩
-
Wynford-Thomas, R., A. Jacob, and V. Tomassini, Neurological update: MOG antibody disease. J Neurol, 2019. 266(5): p. 1280-1286. ↩
-
Chalmoukou, K., et al., Anti-MOG antibodies are frequently associated with steroid-sensitive recurrent optic neuritis. Neurol Neuroimmunol Neuroinflamm, 2015. 2(4): p. e131. ↩
-
Chen, H., et al., Comparisons of the efficacy and tolerability of mycophenolate mofetil and azathioprine as treatments for neuromyelitis optica and neuromyelitis optica spectrum disorder. Eur J Neurol, 2017. 24(1): p. 219-226. ↩
-
Romeo, A.R. and B.M. Segal, Treatment of neuromyelitis optica spectrum disorders. Curr Opin Rheumatol, 2019. 31(3): p. 250-255. ↩
-
Pittock, S.J., et al., Eculizumab in Aquaporin-4-Positive Neuromyelitis Optica Spectrum Disorder. N Engl J Med, 2019. 381(7): p. 614-625. ↩
-
Selmaj, K. and I. Selmaj, Novel emerging treatments for NMOSD. Neurol Neurochir Pol, 2019. 53(5): p. 317-326. ↩
Nik Krajnc, dr. med.
Klinični oddelek za bolezni živčevja,
Nevrološka klinika,
Univerzitetni klinični center Ljubljana
Recenzent:
Jožef Magdič, dr.med.
Oddelek za nevrološke bolezni
Univerzitetni klinični center Maribor
Sprejeto: 3.7.2020
Objavljeno: 3.8.2020