Spletna revija za znanstvenike, strokovnjake
in nevroznanstvene navdušence
Naslovnica Članki Intervjuji Mnenja Zdravje Korenine eSinapsa Številke ![]()
Konformacijske bolezni
članki
eSinapsa, 2011-1
Zvezdan Pirtošek
Eksoskeleti – inteligentne bionske naprave
Marko Munih
O aktualnih dilemah draženja globokih možganskih struktur pri obsesivno - kompulzivni motnji
Nadja Jarc
Sledite svojo srečo ... z iPhone
Urban Kordeš
eSinapsa, 2011-2
Renata Salecl
Gašper Tkačik
Astrociti – spregledane zvezde nevrobiologije
Marko Kreft, Robert Zorec
Sašo Dolenc
Meditacija - malo truda, veliko koristi
Luka Dimic
eSinapsa, 2011-3
Mara Bresjanac
Martina Starc
Rok Berlot
Varnost uporabe generičnih protiepileptičnih zdravil
Mojca Kržan, Matevž Kržan
Možgani, računalniki - nekaj vmes
Miha Pelko
eSinapsa, 2012-4
Ali so moški in ženski možgani različni?
Gregor Majdič
O kognitivnih motnjah pri bolnikih s Parkinsonovo boleznijo
Dejan Georgiev
Akutno možgansko kap lahko uspešno zdravimo
Nina Vujasinovič, Bojana Žvan
Vloga nevropsihološke diagnostike pri odkrivanju zgodnjih znakov alzheimerjeve bolezni
Simon Brezovar
eSinapsa, 2013-5
Srečanje dveh velikanov: možganov in imunskega sistema
Matej Markota
Novo odkritje na področju sporadičnih prionskih bolezni
Jana Jerše, Nadja Jarc
Učinek placeba brez lažnih zdravil in zavajanja
Mara Bresjanac
Subarahnoidna krvavitev zaradi tromboze venskih sinusov
Mateja Repar, Anita Resman Gašperčič
eSinapsa, 2013-6
Odstranjevanje možganskih tumorjev pri budnem bolniku
Andrej Vranič, Jasmina Markovič, Blaž Koritnik
Zmedena bolnica, ki nič ne vidi ali PRES
Manja Hribar, Vid Zgonc
Manja Hribar
Netravmatska lokalizirana konveksitetna subarahnoidna krvavitev
Mateja Repar, Fajko F. Bajrović
Sistemska skleroza in ishemična možganska kap - vzročna povezanost ali le koincidenca?
Mateja Repar, Janja Pretnar Oblak
Klemen Grabljevec
Z omejevanjem spodbujajoča terapija pri bolnikih po nezgodni možganski poškodbi
Dejana Zajc, Klemen Grabljevec
eSinapsa, 2014-7
Možgani v mreži navezanosti, ki nas zaznamuje
Barbara Horvat
Vpliv senzoričnega dotoka na uglasitev možganskih povezav
Peter Gradišnik
Človeški konektom ali kakšne so zveze v naših možganih
Blaž Koritnik
Niko Lah
Torkove delavnice za osnovnošolce
Mateja Drolec Novak, Vid V. Vodušek
Da ne pozabim! Tehnike za pomladitev spomina
Klara Tostovršnik, Hana Hawlina
Površina socialne nevroznanosti
Manuel Kuran
Clarity - bistri možgani Karla Deisserotha
Gregor Belušič
Barbara Gnidovec Stražišar
Bojana Žvan
Nevroplastičnost po možganski kapi
Marjan Zaletel
Klinično psihološka obravnava pacientov po možganski kapi in podpora pri vračanju na delovno mesto
Barbara Starovasnik Žagavec
Možgani: organ, s katerim ljubimo
Andraž Matkovič
Marija Šoštarič Podlesnik
Gibalno-kognitivna vadba: praktična delavnica
Mitja Gerževič, Marina Dobnik
Anton Grad
Nevrologija, imunologija, psihiatrija …
Bojan Rojc
Andraž Stožer, Janez Bregant
Dominika Novak Pihler
Možganska kap – »kako ostati v omrežju?«
Nina Ozimic
Klara Tostovršnik
eSinapsa, 2014-8
Znotrajžilno zdravljenje možganskih anevrizem
Tamara Gorjanc, Dimitrij Lovrič
Obravnava hladnih možganskih anevrizem
Bojana Žvan, Janja Pretnar Oblak
Ali deklice z Rettovim sindromom govorijo z očmi?
Anka Slana, Urška Slana
Progresivna multifokalna encefalopatija
Urša Zabret, Katarina Šurlan Popovič
Ne ubijaj – poskusi na živalih
Martina Perše
Poizkusi na živalih - za in proti
Simon Horvat
eSinapsa, 2015-9
Kako deluje navigacijski sistem v naših možganih
Simon Brezovar
Vsakodnevno delo slepe osebe / s slepo osebo
Denis Kamnar
Uroš Marušič
Manca Tekavčič Pompe
Toni Pustovrh
Marko Hawlina
Od svetlobe do podobe ali kako vidijo svet naši možgani
Simon Brezovar
Janja Hrastovšek
Zala Kurinčič
Pogledi na mejno osebnostno motnjo
Jerica Radež, Peter Kapš
Uvid kot socialno psihološki fenomen
Vid Vodušek
Uvod v vidno-prostorske funkcije s praktičnimi primeri
Ana Bujišić, Sanja Roškar
eSinapsa, 2015-10
Difuzijsko magnetnoresonančno slikanje
Rok Berlot
Katja Pavšič
Radiološko izolirani sindrom - ali ga moramo poznati?
Matej Vouk, Katarina Šurlan Popovič
Kako izgledajo možgani, ki govorijo več jezikov?
Gašper Zupan
Nov pristop v rehabilitaciji - terapija s pomočjo psa
Mateja Drljepan
Pogled v maternico z magnetnoresonančno preiskavo
Taja Jordan, Tina Vipotnik Vesnaver
Saša Zorjan
Saša Zorjan
Nevroestetika: ko nevroznanost obišče galerijo
Anja Voljavec, Hana Hawlina, Nika Vrabič
Ali so psihogeni neepileptični napadi res psihogeni?
Saška Vipotnik, Gal Granda
Kako nam lahko glasna glasba »vzame« sluh in povzroči tinitus
Nejc Steiner, Saba Battelino
eSinapsa, 2016-11
Mara Bresjanac
Kako ultrazvok odpira pot v možgane
Kaja Kolmančič
Kako je epigenetika spremenila nevroznanost
Metka Ravnik Glavač
Ondinino prekletstvo ali sindrom prirojene centralne hipoventilacije
Katja Pavšič, Barbara Gnidovec Stražišar, Janja Pretnar Oblak, Fajko F. Bajrović
Zika virus in magnetnoresonančna diagnostika nepravilnosti osrednjega živčevja pri plodu
Rok Banko, Tina Vipotnik Vesnaver
Motnje ravnotežja otrok in odraslih
Nejc Steiner, Saba Battelino
eSinapsa, 2016-12
Vloga magnetnoresonančne spektroskopije pri obravnavi možganskih tumorjev
Gašper Zupan, Katarina Šurlan Popovič
Tiskanje tridimenzionalnih modelov v medicini
Andrej Vovk
Aleš Oblak
Kevin Klarič
Sinestezija: umetnica, ki ne želi odrasti
Tisa Frelih
Računska psihiatrija: od nevroznanosti do klinike
Nastja Tomat
Kognitivni nadzor: od vsakdanjega življenja do bolezni
Vida Ana Politakis
eSinapsa, 2017-13
Internet: nadgradnja ali nadomestek uma?
Matej Perovnik
Vloga črevesnega mikrobioma pri odzivu na stres
Vesna van Midden
Stres pušča posledice tako na človeškem kot živalskem organizmu
Jasmina Kerčmar
Prikaz normalne anatomije in bolezenskih stanj obraznega živca z magnetno resonanco
Rok Banko, Matej Vrabec
Psihedelična izkušnja in njen zdravilni potencial
Anja Cehnar, Jona Basle
Vpliv hiperglikemije na delovanje možganov
Jasna Šuput Omladič, Simona Klemenčič
Nevrofibromatoza: napredujoče obolenje centralnega in perifernega živčevja
Nejc Steiner, Saba Battelino
Fenomen žrtvenega jagnja v dobi interneta
Dolores Trol
Tesnoba staršev in strategije spoprijemanja, ko pri otroku na novo odkrijejo epilepsijo
Daša Kocjančič, Petra Lešnik Musek, Vesna Krkoč, David Gosar
eSinapsa, 2017-14
Zakaj ne zapeljem s ceste, ko kihnem?
Anka Slana Ozimič, Grega Repovš
Nobelova nagrada za odkritje molekularnih mehanizmov nadzora cirkadianih ritmov
Leja Dolenc Grošelj
Možgani pod stresom: od celic do duševnih motenj
Nastja Tomat
Na sledi prvi vzročni terapiji Huntingtonove bolezni
Danaja Metul
Razlike med spoloma pri Parkinsonovi bolezni
Kaja Kolmančič
eSinapsa, 2018-15
Susceptibilno poudarjeno magnetnoresonančno slikanje pri bolniku z ALS
Alja Vičič, Jernej Avsenik, Rok Berlot
Sara Fabjan
Reverzibilni cerebralni vazokonstrikcijski sindrom – pot do diagnoze
Maja Cimperšek, Katarina Šurlan Popovič
Liam Korošec Hudnik
Kognitivno funkcioniranje pri izgorelosti
Marina Horvat
eSinapsa, 2019-16
Maša Čater
Saša Koprivec
Infekcije osrednjega živčnega sistema s flavivirusi
Maja Potokar
Raziskava: Kako depresija vpliva na kognitivne sposobnosti?
Vida Ana Politakis
Razvoj depresije pri otrocih z vidika navezovalnega vedenja
Neža Grgurevič
Sonja Prpar Mihevc
Umetno inteligentna nevroznanost: srečanje nevronskih mrež in možganske fiziologije
Kristijan Armeni
Čebelji strup pri preventivi nevrodegenerativnih bolezni in priložnost za klinično prakso
Matjaž Deželak
eSinapsa, 2019-17
IgG4+ – skupni imenovalec diagnoz iz preteklosti
Cene Jerele, Katarina Šurlan Popovič
Nov molekulski mehanizem delovanja ketamina v astrocitih
Matjaž Stenovec
Praktični pristop k obravnavi utrujenosti in motenj spanja pri bolnikih z multiplo sklerozo
Nik Krajnc, Leja Dolenc Grošelj
Jure Pešak
eSinapsa, 2020-18
Bolezni spektra anti-MOG pri odraslih
Nik Krajnc
Samomor pod lupo nevroznanosti
Alina Holnthaner
eSinapsa, 2020-19
Ob mednarodnem dnevu znakovnih jezikov
Anka Slana Ozimič
Teorija obetov: kako sprejemamo tvegane odločitve
Nastja Tomat
Sara Fabjan
Matjaž Deželak
Nina Stanojević, Uroš Kovačič
Od človeških nevronov do možganskih organoidov – nova obzorja v nevroznanosti
Vesna M. van Midden
Splošna umetna inteligenca ali statistične jezikovne papige?
Kristijan Armeni
Zunajcelični vezikli kot prenašalci zdravilnih učinkovin preko krvno-možganske prepreke
Saša Koprivec
Matjaž Deželak
eSinapsa, 2021-20
Migrena: starodavna bolezen, sodobni pristopi k zdravljenju
Eva Koban, Lina Savšek
Zgodnji razvoj socialnega vedenja
Vesna Jug
Nastja Tomat
Mikrosplet: povezovanje preko mikrobioma
Tina Tinkara Peternelj
Stimulacija možganov kot način zdravljenja depresije
Saša Kocijančič Azzaoui
eSinapsa, 2021-21
eSinapsa, 2022-22
Sodobni vidiki motenj hranjenja
Karin Sernec
Ples in gibalni dialog z malčki
Neva Kralj
Atul Gawande
Jezikovna funkcija pri Alzheimerjevi bolezni
Gašper Tonin
Dostava terapevtikov preko krvno-možganske pregrade
Matjaž Deželak
eSinapsa, 2022-23
Akutni ishemični infarkt hrbtenjače pri zdravih otrocih – kaj lahko pove radiolog?
Katarina Šurlan Popovič, Barbara Šijaković
eSinapsa, 2023-24
Možganska omrežja pri nevrodegenerativnih boleznih
Tomaž Rus, Matej Perovnik
Morske živali kot navdih za nevroznanstvenike: morski konjiček, morski zajček in klobučnjak
Tina Bregant
Metoda Feldenkrais: gibanje in nevroplastičnost
Mateja Pate
Etično naravnana animalna nevroznanost
Maša Čater
Helena Motaln, Boris Rogelj
eSinapsa, 2023-25
Urban Košak, Damijan Knez, Anže Meden, Simon Žakelj, Jurij Trontelj, Jure Stojan, Maja Zakošek Pipan, Kinga Sałat idr.
eSinapsa, 2024-26
Naravno okolje kot vir zdravja in blagostanja
Karin Križman, Grega Repovš, Gaja Zager Kocjan, Gregor Geršak
Katja Peganc Nunčič, Damjan Osredkar
Tanja Goltnik
Ali je zgodnje vstajanje dedno?
Cene Skubic, Laura Plavc, Damjana Rozman, Leja Dolenc Grošelj
eSinapsa, 2024-27
Širša terapevtska uporaba ketamina: potenciali in izzivi
Kristian Elersič
Moč vpliva socialne opore na bolečino
Jana Verdnik
Benjamin Bušelič
Urška Černe, Anemari Horvat, Robert Zorec, Nina Vardjan
eSinapsa, 2025-28
Maša Čater, Nastja Blagovič, Urška Jerič, Agata Kokalj Malovrh, Nuša Balen, Tanja Kunej
Vpliv izobrazbe na skrb za zdravje možganov
Hana Kos, Matej Perovnik
Selena Horvat, Anja Pišlar
eSinapsa, 2025-29
Knjiga Kje so moji ključi kot primer narativne medicine
Zdenka Čebašek-Travnik, Saša Novak
Vpliv anestetikov na oksidativni stres
Katerina Tomsič, Alenka Nemec Svete
Rak, živčni in imunski sistem – kako so med seboj povezani?
Maja Čemažar, Tanja Jesenko, Urša Lampreht Tratar, Maša Omerzel
eSinapsa, 2026-30
Denis Štepihar, Klementina Fon Tacer
Marta Macedoni Lukšič
eSinapsa, 2026-31
Ste se morda vprašali “Kaj se je zgodilo z boleznijo norih krav?” Po vznemirjenju, ki so ga ob koncu prejšnjega stoletja sprožili prioni, ko so po bolezni pri kravah povzročili tudi epidemijo t.i. variante Creutzfeldt Jakobove bolezni pri ljudeh, o teh boleznih v medijih skorajda ni več slišati. A ne zato, ker bi ne bile aktualne. Raziskave so razkrile, da so nekatere pogoste bolezni, kot je denimo Alzheimerjeva bolezen, presenetljivo podobne prionskim po mehanizmih nastanka, kar odpira nove diagnostične in terapevtske možnosti.
 Medicina razvršča bolezni po različnih kriterijih, od tega, kateri organ(ski sistem) je primarno prizadet, do tega, katera stroka je primarno udeležena v zdravljenju ali lajšanju simptomov bolezni. Ko gre za motnje v delovanju možganov, imamo tako opravka z nevrološkimi na eni in psihiatričnimi stanji na drugi strani. S poglabljanjem razumevanja patofiziologije se tovrstne razmejitve postopno umikajo. Namesto njih postajajo kriterij za nove razvrstitve in temelj za razvoj diagnostičnih in terapevtskih pristopov patogenetski mehanizmi, ki so sorodni sicer navidez zelo raznolikim boleznim.
Medicina razvršča bolezni po različnih kriterijih, od tega, kateri organ(ski sistem) je primarno prizadet, do tega, katera stroka je primarno udeležena v zdravljenju ali lajšanju simptomov bolezni. Ko gre za motnje v delovanju možganov, imamo tako opravka z nevrološkimi na eni in psihiatričnimi stanji na drugi strani. S poglabljanjem razumevanja patofiziologije se tovrstne razmejitve postopno umikajo. Namesto njih postajajo kriterij za nove razvrstitve in temelj za razvoj diagnostičnih in terapevtskih pristopov patogenetski mehanizmi, ki so sorodni sicer navidez zelo raznolikim boleznim.
Primer je skupina konformacijskih bolezni (preglednica 1). Tako sta jih poimenovala1, ki sta jih opredelila kot stanja, do katerih pride, ko neka beljakovina iz svoje nativne, funkcionalne oblike spremeni konformacijo tako, da njene molekule zavzamejo obliko (konformacijo) beta-nagubanega lista in se pričnejo zlagati ena na drugo. Tako nastanejo strukturno zelo urejeni, netopni, vlaknati polimeri, ki imajo pomembne skupne lastnosti ne glede na to, iz katere beljakovine so nastali. Generično ime za te polimere je amiloid, kar izpostavlja njihove skupne ultrastrukturne, biokemijske in histokemijske lastnosti: tvorijo jih nerazvejana vlakna premera 7,5 do 10 nm, amiloidne odlage so kongofilne (obarvajo se z barvilom Kongo rdeče) in v polarizacijskem mikroskopu kažejo zeleno dvolomnost 2. Odlage amiloida so značilnost vseh konformacijskih bolezni, ki pa se razlikujeje po tipu odlag (npr. pri Alzheimerjevi bolezni najdemo beta amiloid v plakih med celicami, beljakovina tau pa se odlaga v obliki pentelj znotraj celic), po tem, v katerih tkivih in organih se odlaga amiloid in po klinični sliki bolezni. Za ilustracijo izpostavimo le nekatere primere raznolikih bolezni, ki jim je skupna konformacijska patologija: sladkorna bolezen tip 2, cistična fibroza, dedni panlobularni emfizem, Alzheimerjeva in Parkinsonova bolezen.
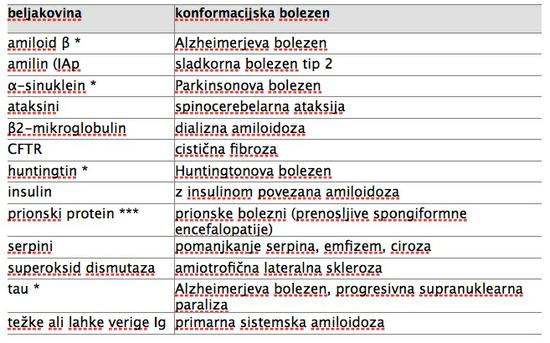
Preglednica 1: Primeri konformacijskih bolezni in beljakovin, ki se pri the boleznih odlagajo. Ena zvezdica * označuje beljakovine, ki so prenosljive na molekularni ravni (t.i. prionoidi), tri zvezdice *** pa označujejo prion, primer bolezensko spremenjene beljakovine, ki je potrjeno kužna po vnosu v organizem z uživanjem hrane s prioni, s presaditvijo organov, invazivnimi posegi v živčevje, s transfuzijo krvi ter eksperimentalno celo po vdihavanju aerosolov s prioni.6
Konformacijska sprememba, ki je podlaga bolezenskemu združevanju in odlaganju beljakovin, naj bi bila prisotna tudi v normalnih okoliščinah kot prehodno in reverzibilno stanje proteinske homeostaze – proteostaze3 (slika 1). Verjetnost nastanka bolezni se poveča, kadar se ravnovesje med nastajanjem in razgradnjo beljakovine prevesi v smer kopičenja. Pri tem lahko konformacijska sprememba povzroči izgubo funkcije nativne beljakovine (tak primer je izguba funkcije transmembranske beljakovine CFTR pri cistični fibrozi). Po drugi strani bolezensko zvita beljakovina lahko pridobi novo, toksično funkcijo (npr. amiloid beta pri Alzheimerjevi bolezni), ali pa v nastanku tkivne okvare in klinične bolezni sodelujeta izguba normalne in pridobitev toksične funkcije.
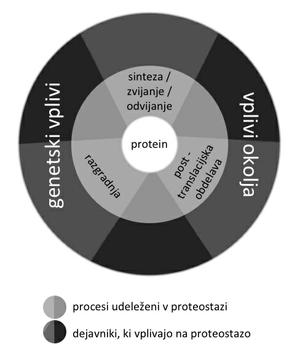
Slika 1: Homeostaza proteinov – proteostaza – so procesi sinteze, post-translacijske obdelave in razgradnje beljakovin, ki vzdržujejo njihovo normalno koncentracijo in delovanje. Genetski dejavniki ter okolje, ki spreminja razmere v organizmu (npr. stres, kalorični vnos …), vplivajo na proteostazo neposredno in posredno.
Okoliščine, ki rušijo proteostazo, so lahko prirojene, kot denimo genske mutacije ter polimorfizmi, ki vplivajo na stabilnost in zvijanje beljakovin, ali pa gre za različne vplive okolja na organizem. Poleg vnetnih procesov, oksidativnega stresa in presnovnih dejavnikov, ki lahko olajšajo in pospešijo razvoj konformacijskih bolezni, lahko sem štejemo še njihovo prenosljivost: bolezensko preobrazbo lahko sproži vnos patološko spremenjene beljakovine v predhodno zdrav organizem. Ker je ves patogeni potencial vsebovan le v strukturnih značilnostih, t.j. v konformaciji beljakovine, pri teh boleznih niso izpolnjeni temeljni Kochovi kriteriji, da bi jih lahko opredelili za “nalezljive”. Prehod nativne beljakovine v bolezensko konformacijo po stiku z patološko beljakovino je v nekaterih primerih dokumentirana le na molekularni ravni (npr. pri amiloidu beta4 in alfa-sinukleinu5). Za razliko od prionov, ki lahko povzročijo bolezen po katerikoli poti vstopanja v organizem6, se zato za amiloid beta, alfa-sinuklein, tau in druge amiloidogene beljakovine, ki so prenašalci konformacijske patologije le ob neposrednem vnosu v dovzetni organ ali tkivo, uveljavlja ime prionoidi7.
Namesto, da bi bile prionske bolezni pozabljene, so torej prav raziskave prionov prispevale k boljšemu razumevanju patofiziologije cele vrste konformacijskih bolezni, odprl pa se je tudi boljši vpogled v primere v biologiji, ki kažejo, da so konformacijske spremembe z nastankom amiloida v določenih razmerah del normalne fiziologije. Primer za fiziološke amiloide je npr. oblikovanje dologoročnih spominskih sledi v aktivnih sinapsah8 prek trajne konformacijske spremembe ustreznih sinaptičnih beljakovin ali denimo skladiščenje hormonov9.
Čeprav konformacijskih bolezni danes še ne znamo zdraviti, boljše razumevanje mehanizma njihovega nastanka odpira pomembne nove smeri za njihovo zgodnjo prepoznavo in razvoj zdravljenja.
-
___
-
Carrell RW, Lomas DA. Conformational disease. Lancet 1997; 350: 134 - 8. ↩
-
Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Eng J Med 2003;349:583-96. ↩
-
Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem 2009; 78:959-91. ↩
-
Kane MD, Lipinski WJ, Callahan MJ, Bian F, Durham RA, Schwarz RD, Roher AE, Walker LC. Evidence for seeding of beta -amyloid by intracerebral infusion of Alzheimer brain extracts in beta -amyloid precursor protein-transgenic mice. J Neurosci 2000; 20, 3606–11. ↩
-
Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synu- clein. Proc Natl Acad Sci USA 2009; 106, 13010–5. ↩
-
Haybaeck J, Heikenwalder M, Klevenz B, Schwarz P, Margalith I, Bridel C, Mertz K, Zirdum E, Petsch B, Fuchs TJ, Stitz L, Aguzzi A. Aerosols Transmit Prions to Immunocompetent and Immunodeficient Mice. PLoS Pathog 7(1): e1001257. doi:10.1371 journal.ppat.1001257. ↩
-
Aguzzi A. Cell biology: Beyond the prion principle. Nature 2009; 459, 924–925. ↩
-
Si K, Lindquist S, Kandel ER. A Neuronal Isoform of the Aplysia CPEB Has Prion-Like Properties. Cell 2003; 115: 879-9. ↩
-
Greenwald J, Riek R. Biology of Amyloid: Structure, Function, and Regulation. Structure 2010; 18: 1244-60. ↩
prof. dr. Mara Bresjanac
Laboratorij za regeneracijo in plastičnost živčevja
Inštitut za patološko fiziologijo, Medicinska fakulteta Univerze v Ljubljani